Las enfermedades neurodegenerativas con acúmulo de hierro cerebral (NBIA en inglés, ENACH en español) son trastornos genéticos (hereditarios) en los que existe una acumulación excesiva de hierro a nivel de los ganglios basales (principalmente el globo pálido) y la sustancia negra, causando un defecto en su funcionamiento.
¿QUÉ IMPORTANCIA TIENE EL HIERRO PARA NUESTRO ORGANISMO/CEREBRO?
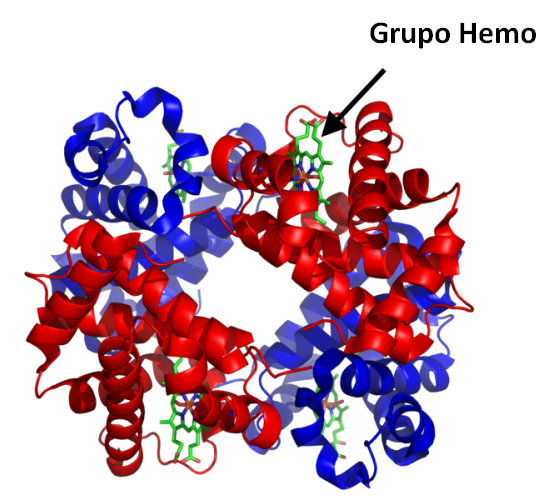
El hierro es un metal esencial para nuestro organismo. Cumple diversas e importantes funciones fisiológicas. La forma en que nuestro organismo lo obtiene es a través de los alimentos. En la sangre forma parte de la hemoglobina (en el grupo Hemo, ver figura), que es la estructura que transporta el oxígeno en los glóbulos rojos para ser distribuido a los distintos tejidos. Es tan importante que nuestro organismo ha desarrollado estrategias para conservarlo. Así, el excedente de hierro se almacena en médula ósea, hígado y bazo. La carencia de hierro produce una disminución de la hemoglobina, lo que llamamos anemia. Su exceso lleva a la acumulación anormal en distintos órganos. El hierro está implicado en procesos vitales como el desarrollo neuronal, la expresión de genes, la función de proteínas (enzimas), la síntesis de neurotransmisores como la dopamina y cumple un rol fundamental en la cadena respiratoria mitocondrial (proceso que permite a las células obtener energía). Dentro del cerebro el hierro se concentra principalmente a nivel de los ganglios basales. Se observa acúmulo de hierro como parte normal del envejecimiento. Cuando las concentraciones de hierro son excesivas se vuelve tóxico porque aumenta el estrés oxidativo celular lesionando proteínas, lípidos y ADN.
¿QUÉ SON LOS GANGLIOS BASALES?
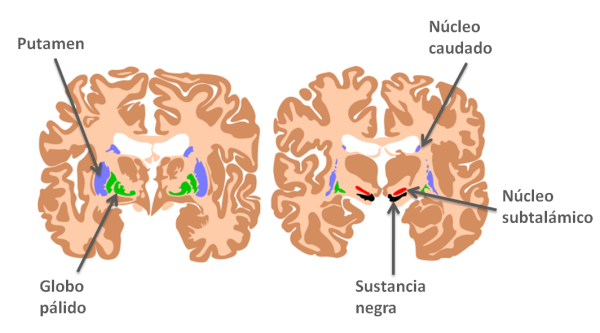
Los ganglios basales son grandes estructuras neuronales (agrupaciones de neuronas) ubicadas en la profundidad de los hemisferios cerebrales. Están formados por el Núcleo Caudado, el Putámen, el Globo Pálido, el Núcleo Subtalámico y la Sustancia Negra. Cumplen una función fundamental en el control motor, emocional y cognitivo, por eso es habitual que las personas que padecen enfermedades que lesionan los ganglios basales presentan alteraciones en alguna de estas tres áreas. PRINCIPALES ENACH Los dos síndromes principales son la neurodegeneración asociada a la pantotenato kinasa (PKAN) y la neurodegeneración asociada PLA2G6 (PLAN). Además existen otros genes menos frecuentes que pueden dar una sintomatología similar y, por otro lado, pacientes con una sintomatología típica sin causa genética identificable (formas “idiopáticas”).
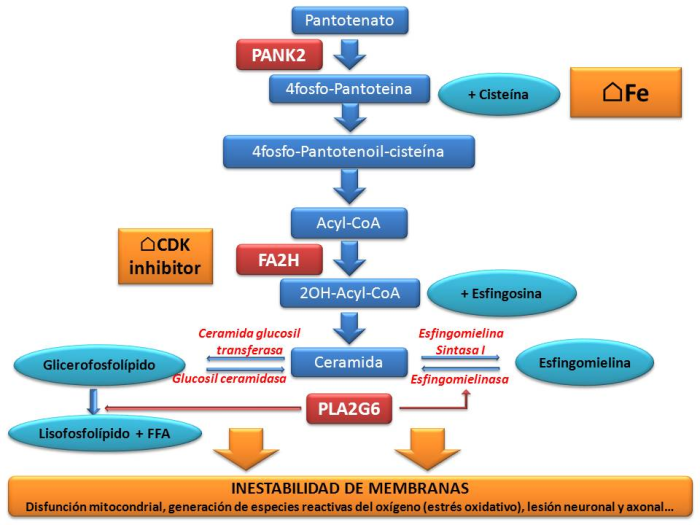
Las ENACH se presentan en la infancia como un trastorno motor progresivo. Por ejemplo pueden comenzar con problemas en la marcha o movimientos anormales tipo corea, distonía, parkinsonismo y espasticidad y/o alteraciones neuropsicológicas. La fisiopatología no es del todo conocida, pero se sabe que la mayoría de los defectos genéticos de este grupo de enfermedades afectan el metabolismo de los fosfolípidos. Por lo tanto, aunque carezcan de marcador bioquímico, es decir, una sustancia que se pueda medir o detectar en la sangre para facilitar su diagnóstico, pueden considerarse enfermedades metabólicas.
¿QUÉ SIGNIFICA UN ERROR METABÓLICO?
Cuando existe un error en el metabolismo, alguna de las reacciones implicadas en él no se produce con la debida eficacia y distintos procesos se ven afectados, en este caso, el metabolismo de los fosfolípidos.
¿POR QUÉ SE PRODUCE UN DÉFICIT DE LAS DISTINTAS ENZIMAS DE LOS ENACH DE CAUSA GENÉTICA?
Cada una de las reacciones del metabolismo, enzimas, transportadores celulares de nuestro cuerpo está determinada genéticamente (codificada). Todos heredamos de nuestros padres la información correcta o alterada que determina que se realice cada una de ellas. La deficiencia de actividad de una proteína enzimática, transportadora o receptora relacionada con el metabolismo se produce debido a mutaciones (cambios estables y hereditarios) en un gen determinado que codifica al transportador o receptor que no funciona correctamente.
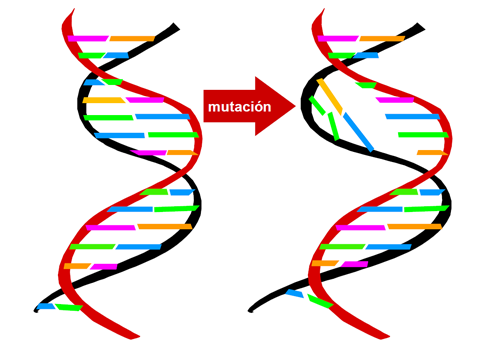
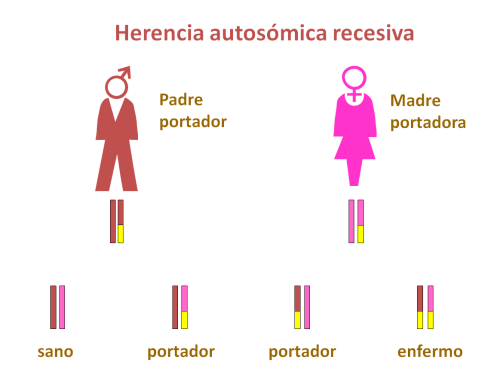
En el caso de las ENACH que se manifiestan en la edad pediátrica, se trata de trastornos genéticos de herencia autosómica recesiva y dominante ligada al cromosoma X. En el primer caso, los padres son portadores de mutaciones en el gen aunque no sufren los efectos de la deficiencia. Si ambos padres transmiten una mutación al niño, éste sufrirá un déficit de causa genética de la proteína codificada. En el segundo caso, se produce una mutación “de novo” en el niño que no estaba presente en los padres. Los genes conocidos hasta el momento son: PANK2, PLA2G6, FA2H, ATP13A2, c19orf12 y WDR45. Las mutaciones en estos genes alterarían las funciones celulares relacionadas con el metabolismo energético, de los neurotransmisores, de fosfolípidos y de ácidos grasos. Estos dos últimos están relacionados con la integridad de las membranas celulares.
¿CÓMO SE REALIZA EL DIAGNÓSTICO DE UNA ENACH?
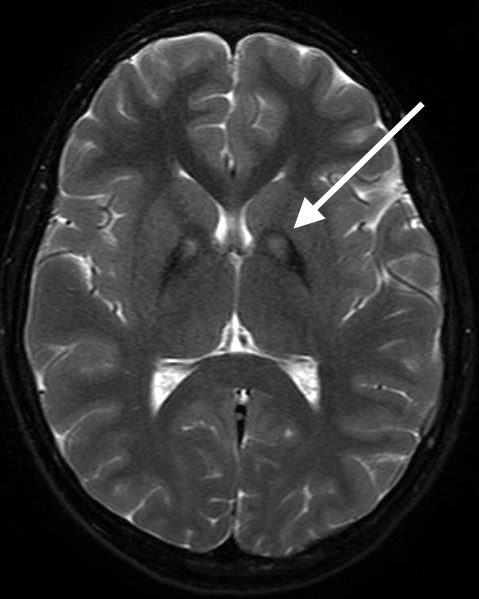
El diagnóstico de estas enfermedades se basa en la presentación clínica y la imagen característica que da la presencia de hierro observada en la resonancia magnética. Dado que este grupo de trastornos pueden asociar afectación a nivel de nervio óptico, la retina, los nervios periféricos y pueden causar crisis epilépticas, suelen ser necesarios otros estudios complementarios para su diagnóstico diferencial. Ante un cuadro de ENACH es necesario realizar un hemograma y determinar el metabolismo del hierro y del cobre (cobre, ceruloplasmina, hierro, ferritina), así como investigar la presencia de acantocitos en una extensión de sangre periférica (PANK2). El examen oftalmológico y los potenciales visuales pueden ser útiles para identificar una retinopatía pigmentaria (PANK2) o una atrofia óptica (PLA2G6 y c19orf12). El estudio periférico puede identificar una neuropatía axonal (PLA2G6 y c19orf12). Finalmente, el diagnóstico específico de cada trastorno se realiza mediante estudios genéticos que evidencian la mutación. Aun así, un porcentaje significativo de casos permanece sin causa genética conocida.
¿QUÉ OCURRE EN EL CASO DE UN NIÑO/NIÑA QUE NACE CON UNA ENACH?
La enfermedad puede debutar a cualquier edad. Dentro de los cuadros clínicos descritos en la edad pediátrica, suele tratarse de niños que nacen normales y tienen un desarrollo normal en los primeros años de vida. En la primera década de la vida debutan las formas clásicas o típicas de distrofia neuroaxonal o PKAN y en la segunda década lo hacen las formas atípicas.
Hay varias causas genéticas que pueden dar lugar a una ENACH generando distintos cuadro clínicos que detallamos a continuación:
1) Neurodegeneración asociada a pantotenato kinasa (PKAN): Es la forma más frecuente de ENACH. Se produce por una mutación en el gen PANK2. Representa aproximadamente la mitad de los casos de ENACH. En la presentación clásica el inicio es temprano (antes de los 6 años) y tiene una progresión rápida. Generalmente comienza con un trastorno de la marcha o la postura con aparición de distonía, presentando aumento del tono muscular (llamado rigidez o espasticidad). El compromiso oromandibular es característico y presentan protrusión lingual severa, problemas de alimentación y para articular las palabras (llamada disartria). Además presentan anomalías a nivel de los movimientos oculares. La evolución es progresiva y el niño afectado puede perder la capacidad de caminar. En la PKAN atípica la presentación es más tardía (13 a 14 años) y la progresión es más lenta. Los trastornos para hablar, caminar y síntomas psiquiátricos pueden presentarse en su inicio. En la mayoría de los casos de PKAN en la RM de cerebro hay un patrón de imagen característico. Corresponde a la acumulación de hierro en globo pálido, que lleva a la descripción del signo de "ojo de tigre".
2) Neurodegeneración asociada a fosfolipasa A2 grupo 6 (PLAN).
3) Neurodegeneración asociada a la hidroxilasa de ácidos grasos (FAHN).
4) Enfermedad de Kufor-Rakeb (PARK9).
5) Neurodegeneración asociada a proteína de la membrana mitocondrial (MPAN).
6) Neurodegeneración asociada a proteína beta-hélice (BPAN).
7) ENACH idiopática.
¿QUÉ HAY QUE HACER PARA EVITAR LAS CONSECUENCIAS DE UNA ENACH?
No hay actualmente un tratamiento curativo para las ENACH. El tratamiento es paliativo de los síntomas de estos trastornos para mejorar la calidad de vida. Los pacientes deben tener un seguimiento multidisciplinario que involucra al pediatra, neurólogo, genetista, oftalmólogo, fisioterapeuta, gastroenterólogo, traumatólogos y neurocirujano, según las necesidades. El tratamiento requiere de medidas básicas, como mantenimiento de un adecuado estado nutricional y vigilar la aparición de trastornos deglutorios (llamada disfagia).
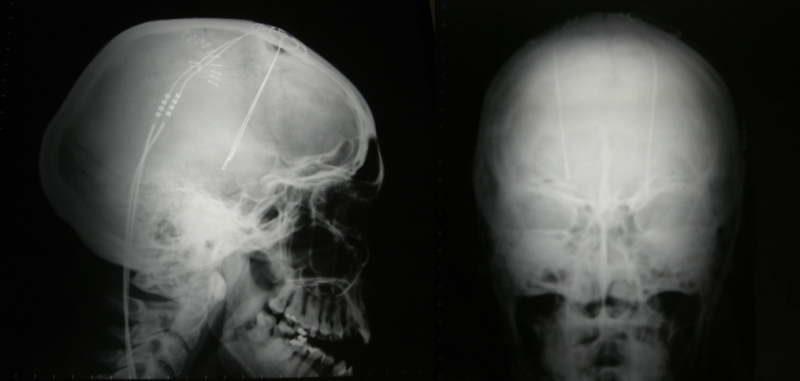
La distonía y la espasticidad pueden ser tratadas con fármacos, como benzodiacepinas, baclofeno y/o trihexifenidilo, así como inyecciones de toxina botulínica en caso de distonías focales. La neurocirugía funcional (estimulación palidal profunda, bomba de baclofeno intratecal) es una estrategia terapéutica en pacientes con distonía generalizada resistentes a fármacos. La estimulación palidal ha sido predominantemente usada en PKAN con mejoría de los espasmos distónicos y de la calidad de vida.
Actualmente hay tratamientos que aún están en fase de investigación, como el deferiprone, un quelante del hierro capaz de atravesar la barrera hemato-encefálica y transferir el hierro a la transferrina circulante desde el compartimento celular. Se ha demostrado que este fármaco puede reducir el depósito de hierro en ganglios basales en pacientes con mutaciones en PANK2, aunque no se ha podido demostrar que estos cambios se traduzcan en una mejoría clínica significativa. Las diferentes enfermedades que causan ENACH son enfermedades hereditarias que, no tratadas, pueden conllevar graves consecuencias. Sin embargo, en muchos casos, el diagnóstico y tratamiento tempranos pueden mejorar la calidad de vida de los pacientes.
Passeig Sant Joan de Déu, 2 08950 Esplugues de Llobregat Barcelona, España Tel: +34 93 203 39 59 www.hsjdbcn.org / www.guiametabolica.org © Hospital Sant Joan de Déu. Todos los derechos reservados.
No hay comentarios:
Publicar un comentario